
The causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic is the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). This highly infectious virus belongs to the coronavirus family. Another virus from the same family is SARS-CoV-1, which is less infectious than SARS-CoV-2. SARS-CoV-1 and SARS-CoV-2 are enveloped, positive-sense, single-stranded RNA viruses which infect the epithelial cells within the lungs.
Scientists have revealed that both the viruses penetrate the host cells via receptor-binding domains (RBDs) of their respective spike proteins that attach to the cell-surface protein angiotensin-converting enzyme 2 (ACE2) of the human host. However, not all the members of the coronavirus family follow the above mechanism to invade the host cells. The initial binding site for Middle East Respiratory Syndrome (MERS) are sialoside binding pocket, found on the cell surface before its attachment to receptor proteins. However, such an interaction in both SARS-CoV and SARS-CoV-2 is not well documented.
From the limited evidence on the primary attachment point of both these viruses, glycosaminoglycans on proteoglycans, such as heparin, could be the initial glycan attachment point. Thereby, this report indicates that sugar-binding sites could be probable binding regions that lie on or close to the RBD. Additionally, scientists have revealed the presence of a region in the spike protein of both SARS-CoV and SARS-CoV-2 that can attach with sialosides. The N-terminal domain (NTD) is also present in SARS-CoV-2-spike that exhibits a putative glycan-binding groove. Scientists believe that it is crucial to understand the role of the host cell surface sialosides in these viruses. Thereby, more research is required to elucidate sugar-binding by SARS-CoV-2-spike protein.
The interaction between putative small-molecule ligands and proteins without the need for labeling/modification of the ligands or proteins, or the need to attach either ligands or proteins to a surface or sensor, is studied using a classical method namely, saturation transfer difference (STD) NMR analysis. In this regard, nuclear spin systems, associated with STD experiments, use saturation pulse to transform the protein from equilibrium to an excited state. Binding of ligand to protein results in two cross-relax, which causes magnetization to pass from the ligand to the protein. Subsequently, in a dynamic binding equilibrium, these two detach, and again the cycle restarts using the saturation pulse.
As 'saturation' cannot be measured, scientists record the pre-set duration and the reduced signal on the ligand. The difference in signal for each resonance from a reference spectrum determines the interaction between a ligand and protein. Even though colloquially, this magnetization transfer is referred to as 'saturation transfer', in reality, this process involves a reduction in signals on the ligands.
The highly modified and complex protein systems are complicated to analyze by the classic STD methods. This is because mammalian proteins often contain large and highly mobile glycans. Glycoproteins may attach to glycans which may cause a spectral overlapping with putative ligand resonances, which results in masking signals.
Another limitation of using classical methods is that the NMR spectra of glycan ligands are overlapping and complex. Additionally, it is possible to implement the STD method only under the condition that the binding of a ligand with a protein is in equilibrium.
A new study posted to the bioRxiv* preprint server focuses on a novel formulation of magnetization or saturation transfer. This method is developed by amalgamating theoretical and computational approaches based on a Bayesian deconvolution algorithm, which helps obtain data more accurately and determine the ligand-protein interaction.
Scientists of the current study revealed that for the "universal' saturation transfer analysis (uSTA), only a series of specific simplified magnetization transfer spectra of a protein-only sample and a variety of ligand concentrations are required. In this analysis, various software is used to generate automatic data on the signal intensity from individual resonances in these spectra. These data can be converted following theoretical treatments. Using these data, scientists have developed structural models of three biologically related ligands, which reveal the consistent presence of sialic acid at the binding region. Hence, uSTA analysis provides complete protein-ligand structures from a raw NMR free induction decay (FID) signal.
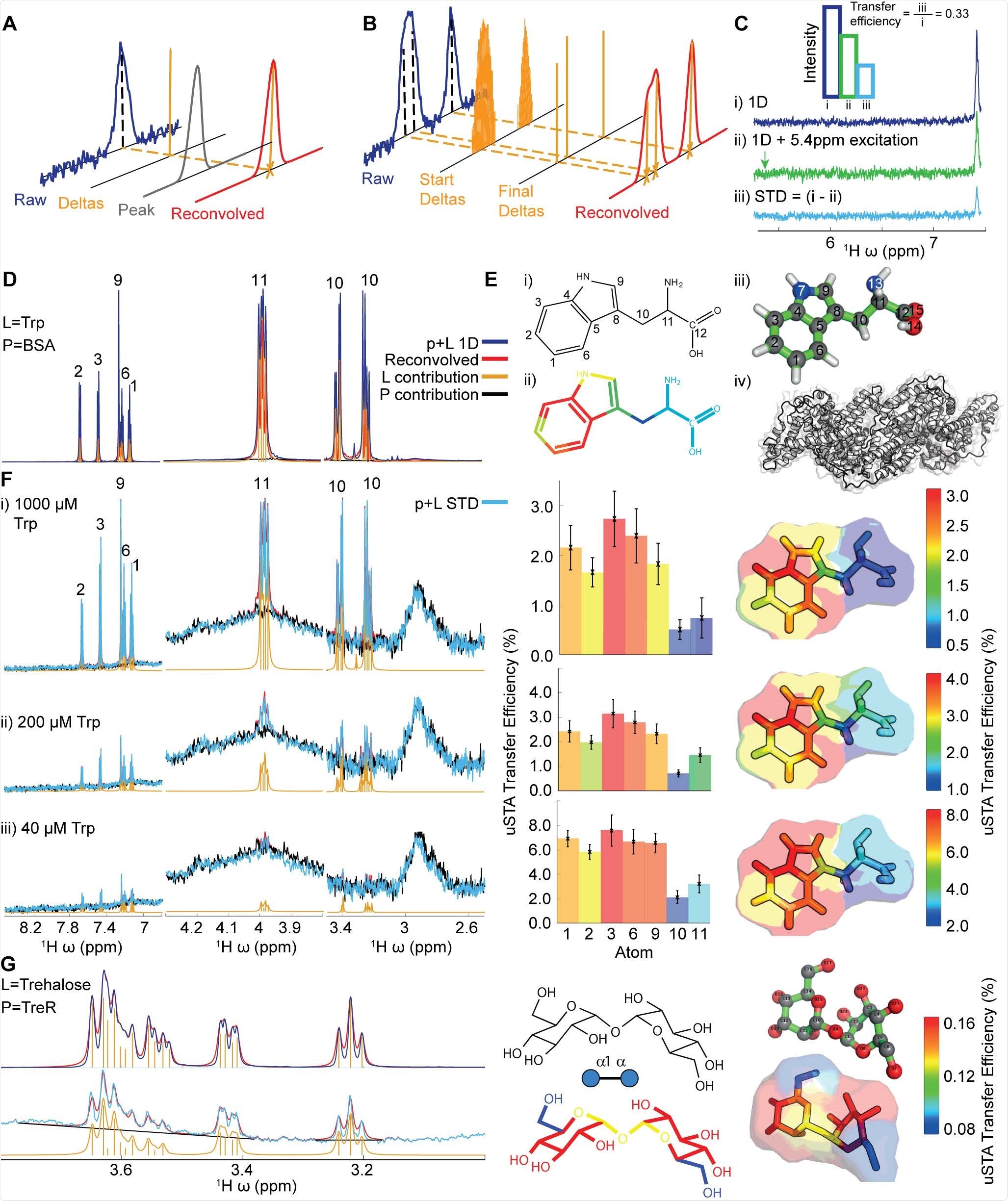
uSTA is used for assessing multiple glycan ligands and effectively identifying intermolecular sugar spike interactions. It helps to identify a glycan-binding site in the NTD of the A-lineage SARS-CoV-2 spike. This study also revealed the importance of cell-surface glycans and the modulation of binding of spike-to-sialosides, in the advancement of the disease and its evolution. uSTA proves extremely critical for mapping direct interactions between natural sialoside sugar ligands and SARS-CoV-2-spike glycoprotein. This is done by quantitating ligand signals in spectral regions which were obscured by resonances from mobile spike-protein glycans.
Scientists found that the genetic modulation of LGALS3BP and B3GNT8 genes could be beneficial for COVID-19 treatment. Additionally, the current research revealed that the A-lineage SARS-CoV-2 virus might have oppressed glycan-mediated attachment to host cells using N-linked-polyLacNAc-chains as a foothold.
To summarize, uSTA offers a comprehensive and quantitative approach for identifying ligands and their binding parameters. It can also identify a binding site for complex, host-derived, and post-translationally modified proteins with putative ligands related to the disease. Thereby, this method is a sophisticated approach that requires minimal inputs and provides multi-faceted analyses of ligand engagement, especially challenging protein systems, such as those exploited by pathogens.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Buchanan, J.C. et al. (2021). Cryptic SARS-CoV2-spike-with-sugar interactions revealed by 'universal' saturation transfer analysis, bioRxiv 2021.04.14.439284; doi: https://doi.org/10.1101/2021.04.14.439284, https://www.biorxiv.org/content/10.1101/2021.04.14.439284v1
Posted in: Medical Research News | Disease/Infection News
Tags: ACE2, Albumin, Angiotensin, Angiotensin-Converting Enzyme 2, Bovine Serum Albumin, Cell, Coronavirus, Coronavirus Disease COVID-19, E. coli, Enzyme, Evolution, Genes, Genetic, Glycan, Glycans, Glycoprotein, Heparin, Ligand, Lungs, Molecule, Pandemic, Protein, Quantitative Analysis, Receptor, Research, Respiratory, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome, Trehalose, Tryptophan, Virus

Written by
Dr. Priyom Bose
Priyom holds a Ph.D. in Plant Biology and Biotechnology from the University of Madras, India. She is an active researcher and an experienced science writer. Priyom has also co-authored several original research articles that have been published in reputed peer-reviewed journals. She is also an avid reader and an amateur photographer.
Source: Read Full Article